Insulin resistance is a characteristic feature of type 2 diabetes.
But although beta cell failure is the ”sine qua non” for development of type 2 diabetes, skeletal muscle insulin resistance is considered to be the initiating or primary defect that is evident decades before beta cell failure and before hyperglycemia develops.
Insulin resistance is defined as a reduced response of target tissues such as skeletal muscle, liver, and adipocytes to insulin.
But skeletal muscle is the predominant site of insulin-mediated glucose uptake in the postprandial state (after a meal).
Around 80% of glucose uptake occurs in skeletal muscle
So studies support the notion that the primary defect in insulin action in patients with T2D resides in the skeletal muscle. Impaired glycogen synthesis in muscle is the primary defect responsible for the insulin resistance.
Genetically it has been shown that if parents have T2D, their child exhibits moderate to severe skeletal muscle insulin resistance despite having normal glucose tolerance (NGT). As they progress from NGT to IGT (impaired glucose tolerance), insulin sensitivity declines markedly but glucose tolerance deteriorates minimally because of a marked insulin secretion. These results, spanning a wide range of ethnic groups, clearly demonstrate that insulin resistance, and not insulin deficiency, initiates the sequence of events leading to the development of T2D. However. progressive beta cell failure is required and ultimately responsible for T2D to become fully manifest.
The normal beta cell response to insulin resistance, irrespective of the etiology of the insulin resistance is to increase its secretion of insulin. However, a chronic physiologic increase in the plasma of insulin concentration has a detrimental effect on skeletal muscle insulin sensitivity. Findings indicate that hyperinsulinemia is not only a compensatory response to insulin resistance but also a self-perpetuating cause of the defect in muscle insulin action.
Muscle lipid accumulation plays a central role in the etiology of the muscle insulin resistance. Fat oxidation is reduced in both T2D and obese insulin resistant nondiabetic individuals, suggesting that muscle mitochondrial oxidative capacity is impaired. It has been shown that NGT children of two T2D parents have a reduced expression of key mitochondrial genes involved in the regulation of oxidative metabolism in skeletal muscle. It is unclear which is the cart and which is the horse: mitochondrial dysfunction leading to increased intramyocellular (inside muscle tissue) lipid content and insulin resistance or increased muscle lipid content (secondary to elevated plasma free fatty acids levels and/or excessive lipid ingestion leading to mitochondrial dysfunction and insulin resistance.
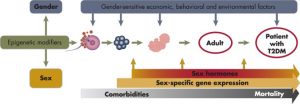
The maintenance of normal glucose homeostasis (ideal glucose metabolism) depends on a finely balanced dynamic interaction between tissue (muscle, liver and fat) sensitivity to insulin and insulin secretion. Even in the presence of severe insulin resistance, a perfectly normal beta cell is capable of secreting sufficient amounts of insulin to offset the defect in insulin action. Thus, the evolution of T2D requires the presence of defects in both insulin secretion and insulin action, and both of these defects can have a genetic as well as an acquired component.
Reference: Skeletal muscle Insulin Resistance Is the Primary Defect in Type 2 diabetes. Ralph A. DeFronzo, MD. Devjit Tripathy, MD. Diabetes Care. 2009 Nov; 32(suppl 2): S157-S163 doi: 10.2337/dc09-S302
{kind=link}